-
Adopt
-
Veterinary Care
Services
Client Information
- What to Expect – Angell Boston
- Client Rights and Responsibilities
- Payments / Financial Assistance
- Pharmacy
- Client Policies
- Our Doctors
- Grief Support / Counseling
- Directions and Parking
- Helpful “How-to” Pet Care
Online Payments
Emergency: Boston
Emergency: Waltham
Poison Control Hotline
-
Programs & Resources
- Careers
-
Donate Now
 By Dan Biros, DVM, DACVO
By Dan Biros, DVM, DACVO
www.angell.org/eyes
ophthalmology@angell.org
617-541-5095
One of the more interesting causes of vision loss in dogs is due to progressive rod cone degeneration (prcd), a hereditary loss of photoreceptor function (often at times mistaken for cataract formation by clients). The disease is caused by a genetic mutation in the photoreceptors of the retina. The cells develop normally, but in adolescence or early adulthood the mutation causes the retinas to degenerate over time leading to blindness.
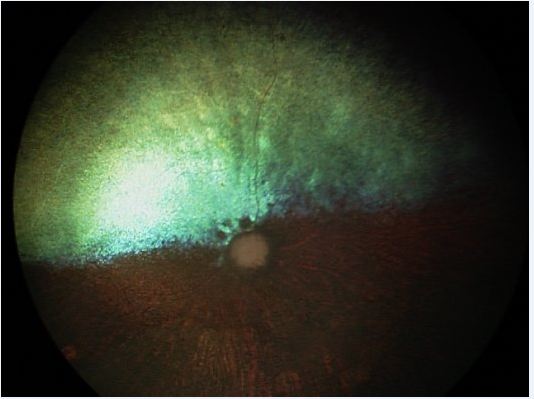 When a patient presents for vision loss and the visual axis is clear, the attention turns to diseases causing retinal dysfunction including glaucoma, sudden acquired retinal degeneration (SARDs), optic neuritis, other CNS disease, retinal dysplasia or detachment, or one of the many varieties of retinal degeneration. The key points of differentiating photoreceptor degeneration from other types of retinal disease is a) the slow rate of progression, b) the characteristic nyctalopia, or night vision loss, as the early indicator of a problem (daytime vision often remains normal for a while), c) the absence of inflammation or ocular hypertension, and d) the often normal appearance to the retina early in the disease. Quite often the eyes are not red, and the pupillary light reflexes (PLRs) may be normal early on with a gradually diminished response as the condition advances. In later stages we will see signs of retinal thinning, namely the retinal blood vessel attenuation and tapetal hyperreflectivity characteristic in the end stage disease (Figure 1). As the retina degenerates over months to years, the optic nerve also atrophies leaving a smaller and often darkened optic disk. By contrast the normal dog fundus has a well-developed retinal vasculature, a large myelinated optic nerve, and a tapetum that is not hyperreflective (Figure 2). Both eyes usually progress equally, but it is not unusual to have unequal PLRs as the degeneration takes place. In later stages the condition may also be associated with cataract development. The relationship of the cataract development to the retinal disease is not clear, but it may be associated with the release of metabolic toxins in the ocular microenvironment that appear as the retina degenerates.
When a patient presents for vision loss and the visual axis is clear, the attention turns to diseases causing retinal dysfunction including glaucoma, sudden acquired retinal degeneration (SARDs), optic neuritis, other CNS disease, retinal dysplasia or detachment, or one of the many varieties of retinal degeneration. The key points of differentiating photoreceptor degeneration from other types of retinal disease is a) the slow rate of progression, b) the characteristic nyctalopia, or night vision loss, as the early indicator of a problem (daytime vision often remains normal for a while), c) the absence of inflammation or ocular hypertension, and d) the often normal appearance to the retina early in the disease. Quite often the eyes are not red, and the pupillary light reflexes (PLRs) may be normal early on with a gradually diminished response as the condition advances. In later stages we will see signs of retinal thinning, namely the retinal blood vessel attenuation and tapetal hyperreflectivity characteristic in the end stage disease (Figure 1). As the retina degenerates over months to years, the optic nerve also atrophies leaving a smaller and often darkened optic disk. By contrast the normal dog fundus has a well-developed retinal vasculature, a large myelinated optic nerve, and a tapetum that is not hyperreflective (Figure 2). Both eyes usually progress equally, but it is not unusual to have unequal PLRs as the degeneration takes place. In later stages the condition may also be associated with cataract development. The relationship of the cataract development to the retinal disease is not clear, but it may be associated with the release of metabolic toxins in the ocular microenvironment that appear as the retina degenerates.
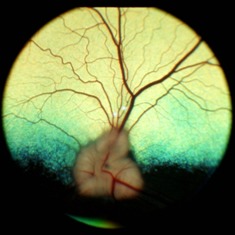
Figure 2: Normal dog fundus
As mentioned prcd does not appear suddenly and can be contrasted to SARDs which is a rapid loss of vision due to loss of retinal function. While most prcd cases are hereditary the SARD’s condition is not. (The cause of SARDs is currently unknown.) The mode of prcd inheritance is usually autosomal recessive, but in select breeds is autosomal dominant (Mastiffs) or even x-linked (Samoyeds and Siberian huskies). Further categories of disease can be further broken down to early- and late onset. Classic early onset prcd can be evident as early as a few weeks of age with late-onset more typically appearing starting at 5-7 years of age. Breeds affected by early onset include Irish setters, Collies, Tibetan terriers, and Cardigan Welsh Corgis. Breeds with late onset disease include miniature and toy poodles, English and American cocker spaniels, English springer spaniels, and Labrador retrievers. Both SARDs and retinal degeneration are confirmed by either the characteristic appearance of the retinas in the later stages or also in early stages by electroretinography. The Optigen independent research laboratory (www.optigen.com), run in consultation with Penn and Cornell researchers, also can provide blood testing for many of the hereditary conditions which has proven helpful in screening for carriers of the disease. This is important since many of the conditions are autosomal recessive. With genetic lineage it can be beneficial to identify carriers and limit propagation of the genetic disease.
There is no cure for prcd and no accepted treatment for slowing the disease or prolonging vision. Causes for the disease have been theorized and among those proposed ideas include changes in plasma lipid levels (docosahexaenoic acid, 22:6n-3). A few veterinary ophthalmologist have developed a neutraceutical product, Ocu-Glo (www.ocuglo.com), containing a mixture of antioxidants and vitamins that may support ocular health in the face of retinal disease. There is no proof that it slows down or reverses the damage caused by the genetic changes, but it may promote the optimal conditions for vision during disease progression. Large randomized studies are needed to support any testimony that the product actually stabilizes or preserved retinal function and vision. We offer this product to clients who want to try something that may help, but we say up front there is no documented medicinal benefit by way of peer reviewed papers at this time.
For more information about Angell’s Ophthalmology service, please visit www.angell.org/eyes. Drs. Coster and Biros are available for consults or referrals at 617 541-5095, or e-mail ophthalmology@angell.org.